Sara Sigüenza González IB
El síndrome MELAS es una de las enfermedades causadas por una mutación en el ADN mitocondrial. Como consecuencia, es de herencia materna y presenta cuadros multisistémicos.
Las mutaciones en el ADN mitocondrial pueden estar localizadas en diferentes lugares: mutaciones que afectan a proteínas de la fosforilación oxidativa, mutaiones en los genes ARN transferentes y mutaciones en los genes que codifican los ARN ribosómicos.
A su vez, las mutaciones en el ADN mitocondrial pueden ser de carácter puntual (mutaciones monogénicas) o bien grandes delecciones de genes.
El síndrome MELAS se encuentra dentro de las enfermedades causadas por mutaciones puntuales en genes codificantes de ARNtransferente (ARNt).
Los ARN de transferencias on los encargados de transportar los aminoácidos que van a formar las proteínas a los ribosomas.Ordenan estos aminoácidos a lo largo de las moléculas ARN mensajeras. Los aminoácidos se unen mediante enlacen peptídicos para formar las proteínas. Hay una molécula ARNt para cada aminoácido, con un triplete de bases complementario conocido como anticodón.
La consecuencia de ello, es que se va a ver afectado el proceso de síntesis de proteínas en los ribosomas mitocondriales.
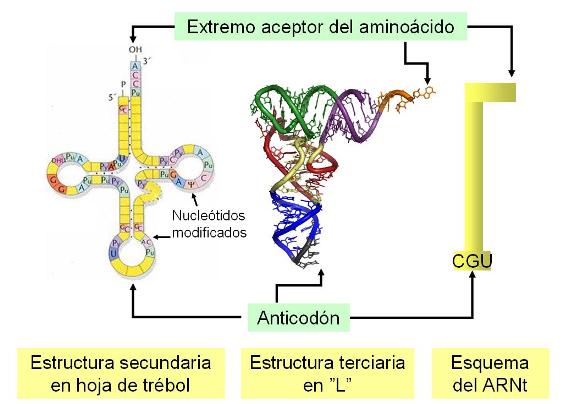
En el genoma mitocondrial encontramos proteínas que intervienen en la síntesis de ATP.
El ADN mitocondrial es una molécula circular de 16 569 pares de bases. Se ha secuenciado y actualmete se sabe que contiene 37 genes:
- 2 de ácido ribonucleico ribosomal (ARNr)
- 22 ARN de transferencia (ARNt)
- 13 polipéptidos que son subunidades de enzimas de los complejos de la fosforilación oxidativa
- 7 subunidades del complejo I
- 1 del complejo III
- 3 del complejo IV
- 2 del complejo V
La fosforilación oxidativa: proceso mediante el cual se obtiene energía ATP en la célula a través del sistema generador OXPHOS, que está compuesto por 5 complejos enzimáticos proteinlipídicos.

Como hemos dicho anteriormente, las mutaciones en los genes que codifican ARNt van a afectar a la síntesis de proteínas codificadas en el genoma mitocondrial. El resultado es un déficit enzimático de varios componentes del sistema OXPHOS; va a tener como consecuencia carencias en el aporte energético.
Uno de los síndromes más comunes debido a mutaciones en genes codificantes de ARNt es la Encefalomiopatía con acidosis láctica y episodios ictus-like (MELAS)
 El síndrome MELAS está causado por la mutación A3243G
El síndrome MELAS está causado por la mutación A3243G
Es una mutación en el gen MT-TL1 del ADN mitocondrial, que pertenece a la familia de los genes de ARN transferente. Este contiene la información para sintetizar una molécula de ARNt denominada ARNtLeu(UUA/G) . Esta molécula se encarga de transportar y ensamblar los aminoácidos de Leucina durante la síntesis proteica mitocondrial.En el 80% de los casos, el síndrome MELAS está causado por una mutación de cambio de adenina por guanina en la posición 3243 del gen MT-TL1. El otro 20% está causado por mutaciones en los siguiente genes: MT-ND1 (gen de la NADH deshidrogenasa I); MT-ND5 (gen de la NADH deshidrogenas IV, mutación G13513A ), MT-TH (gen del ARNtHis ), etc. Estas mutaciones son bastantes más raras en la población que la del gen MT-TL1

Estos genes, proporcionan la información para sintetizar proteínas encargadas en el funcionamiento normal de las mitocondrias, ya que imposibilitan la habilidad de sintetizar energía.
Consecuencia: sintomatología
Como en todas las enfermedades causadas por mutaciones mitocondriales, fisiológicamente se van a ver afectados los tejidos cuyo requerimiento energético es más alto (cerbro, sistema nervioso y músculo...). Este síndrome se caracteriza por encefalomiopatía, acidosis láctica y accidentes vasculares cerebrales de localización preferente parietooccipital que pueden ocasionar hemiparesia, hemianopsia y ceguera cortical. Otras manifestaciones frecuentes son: convulsiones, sordera, migraña, vómitos de repetición, demencia y miopatía. (La sintomatología se desarrolla más ampliamente en el apartado clínico).Los invesigadores no han alcanzado a determinar como los cambios en el genoma mitocondrial llevan a mostrar estos síntomas específicos de MELAS. Las investigaciones se están centrando actualmente en ver los efectos de estas mitocondrias mutadas sobre los tejidos, en especial el tejido nervioso
Patrón de herencia del síndrome MELAS
El síndrome MELA sigue un patrón de heteroplasmia y herencia materna. Ello se debe a que tras la fecundación, las mitocondrias del espermatozoide son eliminadas y solo pasan a formar parte del cigoto las mitocondrias provenientes del óvulo, las maternas. Los trastornos mitocondriales pueden aparecer en todas las generaciones de una familia y en igual manera en hombres que en mujeres. Debido a que se trata de una enfermedad heteroplásmica, algunos individuos pueden haber heredado un menor número de mitocondrias mutadas (ya que el reparto es al azar) y no presentar fenotípicamente la enfermedad. Entre individuos de una misma familia la expresión fenotípica va a variar en función del porcentaje de ADN mitocondrial mutado heredado.Como hemos dicho anteriormente, la herencia de trastornos mitocondriales es materna, por lo que solo las mujeres pasarán la enfermedad a su descendencia; mientras que los varones afectados nunca transmitirán la enfermedad.
Este es un patrón común de mutación mitocondrial:

No hay comentarios:
Publicar un comentario